

Polyarteritis Nodosa
Last updated: November 7, 2014
Synonyms: Polyangiitis, periarteritis nodosa, systemic necrotizing vasculitis
(PAN is part of this larger disease spectrum).
ICD-9 Code: 446.0.
ICD-10 Code: M30.0
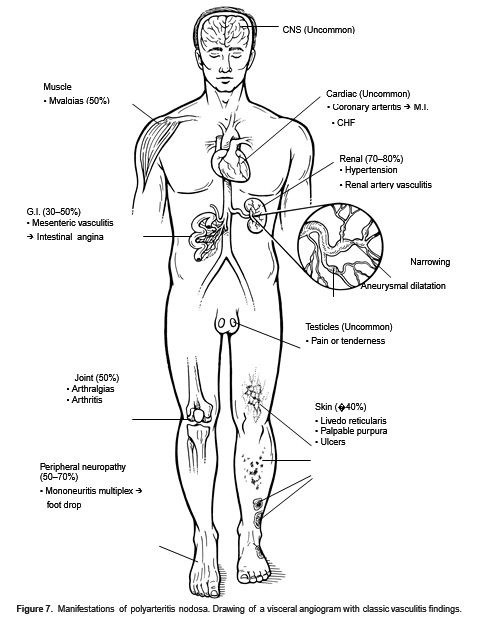
Figure 7. Manifestations of polyarteritis nodosa. Drawing of a visceralangiogram with classes vasculitis findings.
Definition: PAN is a systemic necrotizing vasculitis that affects medium and small muscular arteries of the skin, kidneys, muscle, GI tract, and peripheral nerves. Microscopic polyangiitis is also described because it is a PAN variant and affects smaller sized arteries. Microscopic polyangiitis is distinguished from PAN by manifesting pulmonary capillaritis, alveolar hemorrhage, and P- ANCA positivity.
Etiology: PAN is systemic vasculitis of uncertain etiology. Although there is no consistent infectious cause; PAN has been associated with hepatitis B antigenemia, especially in intravenous substance abusers. Hepatitis C has been described in both PAN and microscopic polyangiitis.
Pathology: Immune complexes deposited in the vascular endothelium activate complement, attracting inflammatory cells. The cellular infiltrate contains both neutrophils and mononuclear cells. Release of proteolytic enzymes and oxygen free radicals results in destruction of the entire vessel wall with fibrinoid necrosis. Eosinophils and granuloma are very rare.
Demographics: PAN and microscopic polyangiitis are uncommon disorders. Median onset age is 40 to 60 years, with a 2:1 male predominance.
Cardinal Findings: Significant constitutional symptoms may predominate early and include fever, weight loss, and severe malaise. Arthralgias/myalgias (50%) and severe abdominal pain often herald disease onset. In PAN, visceral organ manifestations (in order of decreasing frequency) include (Fig. 7):
—Renal (70%–80%): Impaired renal function, proteinuria, and hypertension are secondary to renal artery involvement.
—Nerve (50%–70%): Peripheral neuropathy is common and often manifests early in the disease as mononeuritis multiplex (vasculitis of the vasa nervorum) and may present as foot drop. Distal sensorimotor polyneuropathy and late CNS involvement leading to encephalopathy also occur.
—GI (25%–70%): Severe abdominal pain with intestinal angina is common. GI hemorrhage, perforation, cholecystitis, pancreatitis, intrahepatic aneurysm rupture, and appendicitis have all been reported.
—Cardiac (<10%): Although coronary vasculitis is common at postmortem, symptomatic coronary vasculitis and cardiac disease are uncommon in PAN.
—Skin (40%): Livedo reticularis, nodules, nonspecific maculopapular eruptions, and ulcerations are seen. Subcutaneous skin lesions are typical in the more limited form, cutaneous PAN.
Uncommon Findings: PAN seldom involves the cerebral or pulmonary vasculature. Palpable purpura is also uncommon. In contrast, microscopic polyangiitis often involves the lung and heart.
Complications: Infarction of the bowel leading to hemorrhage and/or perforation is one of the most feared complications. Congestive heart failure and myocardial infarction are occasionally seen. Microscopic polyangiitis can be accompanied by rapidly progressive glomerulonephritis.
Microscopic polyangiitis: Microscopic polyangiitis and PAN share a similar onset and range of signs and symptoms. Microscopic polyangiitis commonly manifests with palpable purpura, alveolar hemorrhage, symptomatic cardiac involvement but is less likely to have hypertension, neuropathy, or hepatitis B. Distinctive pathology includes pulmonary capillaritis, necrotizing segmental glomerulonephritis, and P-ANCA serologies (Table 31). Although the prognosis is good, those with alveolar hemorrhage are at risk of early death.
Diagnostic Testing: Nonspecific inflammatory markers include elevated ESR or CRP, leukocytosis, anemia of chronic disease, and thrombocytosis. In the setting of renal disease, an abnormal urinalysis with proteinuria, granular or red blood cell casts, and declining creatinine clearance is frequently seen. Hypocomplementemia (low C4, C3) and circulating immune complexes are inconsistently detected; the latter are not recommended because of their poor sensitivity and specificity. Evidence of HBV or HCV infection should be sought. An angiogram of the mesenteric or renal arteries may strongly suggest PAN if vascular tapering, microaneurysms, and beading are noted. A histologic diagnosis is made by biopsy of a medium-sized muscular artery. Biopsies are best performed in clinically involved tissues and may include a sural nerve (with foot drop or neuropathy), a muscle (with myopathy or myalgia), or, less preferably, a symptomatic testicle. Renal biopsy is not helpful because specimens seldom exhibit characteristic abnormalities. Microscopic polyangiitis is associated with P-ANCA.
Keys to Diagnosis: PAN may be a diagnostic challenge. Histopathologic evidence of medium-vessel vasculitis or a pathognomonic angiogram in the appropriate clinical setting confirms a clinical diagnosis.
Diagnostic Criteria: The 1990 ACR criteria include (a) weight loss of at least 4 kg; (b) livedo reticularis; (c) testicular pain or tenderness; (d) myalgias, weakness, or leg tenderness; (e) mononeuropathy or polyneuropathy; (f) diastolic blood pressure >90 mm Hg; (g) BUN >40 mg/dL or creatinine >1.5 mg/dL; (h) HBV antigen or antibodies in serum; (i) arteriographic abnormalities (e.g., occlusions or aneurysms); and (j) small- or medium-sized artery biopsy specimen demonstrating PMN leukocytes. The presence of at least three of these features yields an 82% sensitivity and 87% specificity for the diagnosis of PAN.
| Table 31 Comparison if polyarteritis Nodosa and Microscopic Polyangiitis |
||||
| PAN | MPA | |||
|---|---|---|---|---|
| NeuropathyHypertensionAlveolar hemorrhageGlomerular diseaseHepatitis BP-ANCA | 50%-70%CommonAbsentAbsent (vascular nephropathy)Uncommon10% | <35%Uncommon20%-30%Segmental necrotizing glomerulonephritisAbsent80% | ||
|
PAN,polyarteritisnodosa;MPA,microscopicpolyangiitis;P-ANCA,perinuclearantineutrophilcytoplasmic antibody. |
||||
Differential Diagnosis: Alternative forms of systemic vasculitis with overlapping features include granulomatosis with polyangiitis, microscopic polyangiitis, Churg-Strauss angiitis, hypersensitivity vasculitis, and angiitis secondary to amphetamine or cocaine abuse. The vasculitides are compared in Vasculitis: Comparison Table. Medium-vessel arteritis may be a feature of many connective tissue and idiopathic inflammatory disorders (RA, SLE, cryoglobulinemia, Behçet’s syndrome). Occult malignancy, infective endocarditis, cholesterol emboli syndrome, and left atrial myxoma are also in the differential diagnosis. Hairy cell leukemia has been associated with PAN-like picture.
Therapy: In PAN, high-dose corticosteroids are the first line of treatment and are generally begun with at least 1 mg/kg in divided doses. Addition of such cytotoxic agents as cyclophosphamide and less commonly azathioprine is often justified, both for steroid sparing and for disease refractory to steroids alone. Some studies suggest improved survival with the use of cyclophosphamide. MTX has not been studied. Although trials are in progress, TNF inhibitors (infliximab, etanercept) have nonetheless been used with success in some patients with refractory PAN and granulomatosis with polyangiitis. The aggressiveness of therapy is based on the tempo of the disease and the extent of visceral involvement. Addition of plasma exchange to the therapeutic regimen is not of proven benefit. Antiviral therapy with interferon alpha should be considered in patients with PAN with evidence of active HBV infections. The benign course of limited cutaneous PAN supports conservative therapy for this variant.
Prognosis: PAN is associated with very high morbidity and substantial mortality. Untreated, mortality is >90%, but with corticosteroids, it decreases to 50%. The survival advantages of cytotoxic therapy are controversial. Most mortality is owing to renal failure, GI perforation/hemorrhage, and cardiac involvement. Patients who survive the initial manifestations of PAN are at high risk of infectious complications because of the potent immunosuppressive therapy necessary to control the disease. Chronic cardiac and/or renal disease are other long-term sequelae.
BIBLIOGRAPHY
Langford CA. Treatment of polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome: where do we stand? Arthritis Rheum 2001;44:508–512. PMID:11263763
Langford CA. Vasculitis. J Allergy Clin Immunol 2003;111(suppl):S602–S612. PMID:12592306
Lhote F, Guillevin L. Polyarteritis nodosa, microscopic polyangiitis and Churg Strauss syndrome: clinical aspects and treatment. Rheum DisClin North Am 1995;21:911–948. PMID:8592743
Lightfoot RW, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum 1990;33:1088–1093. PMID:1975174
Soto O, Conn DL. Polyarteritis nodosa and microscopic polyangiitis. In: Hochberg MC, Silman AJ, Smolen JS, et al., eds. Rheumatology, 3rd ed. Edinburgh: Mosby, 2003:1611–1621.